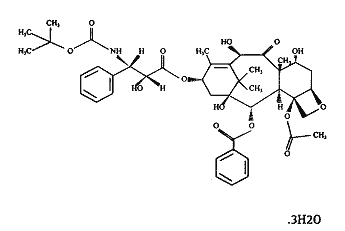
docetaxel
The structural formula is given below:
CAS-148408-66-6
Docetaxel is: (2R,3S)-N-carboxy-3-phenylisoserine, N-tert-butyl ester, 13-ester with 5, 20-epoxy-1, 2, 4, 7, 10, 13-hexahydroxytax-11-en-9-one 4-acetate 2-benzoate, trihydrate.
Docetaxel is a white to almost white powder with the empirical formula C43H53NO14.3H2O and a molecular weight of 861.9. It is highly lipophilic and practically insoluble in water.
Single-dose vials of TAXOTERE concentrated injection containing 20 or 80 mg of docetaxel (anhydrous), in 0.5 or 2.0 mL polysorbate 80, respectively. The sterile pyrogen-free viscous solution contains 40 mg/mL docetaxel (anhydrous). Each mL of docetaxel solution contains 40 mg docetaxel anhydrous and 1040 mg polysorbate 80.
The corresponding vials of diluent for TAXOTERE 20 mg and 80 mg concentrated injections contain 1.98 and 7.33 mL of ethanol respectively in water for injections.
TAXOTERE is an antineoplastic agent which acts by promoting the assembly of tubulin into stable microtubules and inhibits their disassembly which leads to a marked decrease of free tubulin. The binding of docetaxel to microtubules does not alter the number of protofilaments.
TAXOTERE has been shown in vitro to disrupt the microtubular network in cells which is essential for vital mitotic and interphase cellular functions.
TAXOTERE was found to be cytotoxic in vitro against various murine and human tumour cell lines and against freshly excised human tumour cells in clonogenic assays. Docetaxel achieves high intracellular concentrations with a long cell residence time. In addition, TAXOTERE was found to be active on some, but not all, cell lines overexpressing the p-glycoprotein which is encoded by the multidrug resistance gene. In vivo, TAXOTERE is schedule independent and has a broad spectrum of experimental antitumour activity against advanced murine and human grafted tumours. Against transplantable murine tumours in vivo, docetaxel was synergistic with vincristine (administered at the same time), etoposide, cyclophosphamide or 5-fluorouracil, but not with vincristine (administered 24 hours apart), cisplatin or doxorubicin.
The pharmacokinetics of docetaxel have been evaluated in cancer patients after administration of 5-115 mg/m2 in phase I studies. The kinetic profile of docetaxel is dose independent and consistent with a three-compartment pharmacokinetic model with half lives for the α, β and γ phases of 4 minutes, 36 minutes and 11.1 hour, respectively. The initial rapid decline represents distribution to the peripheral compartments and the late phase is due, in part, to a relatively slow efflux of docetaxel from the peripheral compartment. Following the administration of a 100 mg/m2 dose given as a one-hour infusion a mean peak plasma level of 3.7 µg/mL was obtained with a corresponding AUC of 4.6 h.µg/mL. Mean values for total body clearance and steady-state volume of distribution were 21 L/h/m2 and 113 L, respectively.
A study of 14C-docetaxel has been conducted in three cancer patients. Docetaxel was eliminated in both the urine and faeces following oxidative metabolism of the tert-butyl ester group, within seven days, the urinary and faecal excretion account for about 6% and 75% of the administered radioactivity, respectively. About 80% of the radioactivity (60% of the administered dose) recovered in faeces is excreted during the first 48 hours as one major and 3 minor inactive metabolites and very low amounts of unchanged drug.
A population pharmacokinetic analysis has been performed with TAXOTERE in 577 patients. Pharmacokinetic parameters estimated by the model were very close to those estimated from Phase I studies. The pharmacokinetics of docetaxel were not altered by the age or sex of the patient. In a small number of patients (n=23) with clinical chemistry data suggestive of mild to moderate liver function impairment (ALT, AST ≥ 1.5 times the upper limit of normal associated with alkaline phosphatase ≥ 2.5 the upper limit of normal), total clearance was lowered by on average 27% (see Dosage and Administration section). Docetaxel clearance was not modified in patients with mild to moderate fluid retention. No data is available in patients with severe fluid retention.
Docetaxel is more than 95% bound to plasma proteins. Dexamethasone did not affect protein binding of docetaxel.
The effect of prednisone on the pharmacokinetics of docetaxel administered with standard dexamethasone premedication has been studied in 42 patients. No effect of prednisone on the pharmacokinetics of docetaxel was observed.
Phase I studies evaluating the effect of capecitabine on the pharmacokinetics of docetaxel and the effect of docetaxel on the pharmacokinetics of capecitabine showed no effect of capecitabine on the pharmacokinetics of docetaxel (Cmax and AUC) and no effect of docetaxel on the pharmacokinetics of the main capecitabine metabolite 5'DFUR.
Eight phase II studies were conducted in patients with locally advanced or metastatic breast carcinoma. A total of 172 patients had received no prior chemotherapy (previously untreated) and 111 patients had received prior chemotherapy (previously treated) which included 83 patients who had progressive disease during anthracycline therapy (anthracycline resistant). In these clinical trials, TAXOTERE was administered at a 75 mg/m2 dose in 55 previously untreated patients and 100 mg/m2 in 117 previously untreated and 111 previously treated patients. In these trials, TAXOTERE was administered as a one-hour infusion every 3 weeks.
In the intent-to-treat analysis on previously untreated patients, the overall response rate (ORR) was 47% with 9% complete responses (CR). The median duration of response was 34 weeks and the time to progression was 22 weeks.
There was a high response rate in patients with visceral metastases (48.6% in 35 untreated patients).
In patients with ≤ 2 organs involved, the response rate was 58.6% and in patients with ≥ 3 organs involved was 29.4%.
A significant response rate was seen in patients with liver metastases (45% in untreated patients). The same activity is maintained in untreated patients with soft tissue disease (55.5%).
Phase II Trials
In the intent-to-treat analysis on previously untreated patients, the overall response rate (ORR) was 56% with 9.4% complete responses (CR). The ORR was 48.6% with 3.6% CR in the previously treated population including 48.2% ORR with 3.6% CR in the anthracycline resistant patients. The median duration of response was 30 weeks in the previously untreated population, 28 weeks in the previously treated population and 27 weeks in the anthracycline resistant patients. The time to treatment failure was 21 weeks in the previously untreated population, 19 weeks in the previously treated population and 19 weeks in the anthracycline resistant patients.
The 100 mg/m2 dose is associated with higher toxicity.
There was a high response rate in patients with visceral metastases (53.8% in 78 untreated patients, 55.1% in 69 pretreated patients and 53.1% in the subgroup of 49 anthracycline resistant patients).
In patients with ≥ 3 organs involved, the response rate was 54.3 % in previously untreated patients, 55.8 % in previously treated patients and 50 % in the subgroup of anthracycline resistant patients.
A significant response rate was seen in patients with liver metastases (59.5% in untreated patients, 47.2% in previously treated patients and 40% in the subgroup of anthracycline resistant patients). The same activity is maintained in patients with visceral involvement (70.4% in previously untreated, 63.6% in previously treated and 63.2% in the subgroup of anthracycline resistant patients).
Phase III Trials
Two randomised phase III comparative studies, involving a total of 326 alkylating agent failure and 392 anthracycline failure metastatic breast cancer patients, have been performed with docetaxel 100 mg/m2 administered every 3 weeks for seven and ten cycles respectively.
In alkylating agent failure patients, there were no significant differences in median time to progression or median survival between docetaxel ("D"; n=161) and doxorubicin ("DX"; n=165; 75 mg/m2 every 3 weeks) on intent-to-treat and evaluable patient analyses. For the intent-to-treat analysis, median time to progression was 5.9 months for docetaxel and 4.9 months for doxorubicin (D-DX diff: 1.0 months; 95% CI for diff: -0.5 to 1.9); median overall survival was 14.7 months for docetaxel and 14.3 months for doxorubicin (D-DX diff: 0.4 months; 95% CI for diff: -1.9 to 2.7). There was a significant difference in response rates between the two groups: 47.8% for docetaxel and 33.3% for doxorubicin (D-DX diff: 14.5%; 95% CI for diff: 3.9 to 25.0) in intent-to-treat analysis.
In anthracycline failure patients, docetaxel (n=203) was compared to the combination of mitomycin C and vinblastine ("MV"; n=189; 12mg/m2 every 6 weeks and 6mg/m2 every 3 weeks respectively). For the intent-to-treat analysis, docetaxel increased response rate (30% versus 11.6%; D-MV diff: 18.4%; 95% CI for diff: 10.6 to 26.2), prolonged median time to progression (4.3 months versus 2.5 months; D-MV diff: 1.8 months; 95% CI for diff: 1.0 to 2.4) and prolonged median overall survival (11.5 months versus 8.7 months; D-MV diff: 2.8 months; 95% CI for diff: 0.1 to 4.3). Similar results were observed in the evaluable patient analysis.
An open-label, multicentre, randomised phase III study was conducted to compare TAXOTERE and paclitaxel in the treatment of advanced breast cancer in patients where previous therapy should have included an anthracycline. A total of 449 patients were randomised to receive either TAXOTERE 100mg/m2 as a one hour infusion or paclitaxel 175 mh/m2 as a 3 hour infusion. Both regimes were administered every 3 weeks. Efficacy results are described in the following table.
Efficacy of TAXOTERE versus paclitaxel in the treatment of advanced breast cancer (Intent-to-Treat Analysis, unless specified)
| Endpoint | TAXOTERE 100mg/m2 n=225 |
paclitaxel 175mg/m2 n-224 |
p-value (unadjusted) |
|---|---|---|---|
| Median survival (months) 95% CI |
15.3 (13.3 - 18.5) |
12.7 (10.5 - 14.8) |
0.03 |
| Median time to progression (weeks) 95% CI |
24.6 (20 - 30.1) |
15.6 (13.4 - 18.1) |
< 0.01 |
| *Overall response rate (ORR) (%) 95% CI |
32.0 (25.9 - 38.1) |
25.0 (19.3 - 30.7) |
0.10 |
| *ORR in the evaluable population (%) 95% CI |
37.0 (30.2 - 43.9) |
26.0 (19.9-31.9) |
0.01 |
*Primary study endpoint
The most frequent adverse events reported for TAXOTERE were neutropoenia, febrile neutropoenia, gastrointestinal disorders, neurologic disorders, asthenia and fluid retention. More grade 3/4 events were observed from TAXOTERE (55.4%) compared to paclitaxel (23.0%). No unexpected toxicities were reported for TAXOTERE.
TAXOTERE in combination with capecitabine was assessed in an open label, multicentre, randomised trial. A total of 511 patients with locally advanced and/or metastatic breast cancer resistant to, or recurring after an anthracycline containing therapy, or relapsing during or recurring within two years of completing an anthracycline containing adjuvant therapy were enrolled. In this trial, 255 patients were randomised to receive capecitabine (1250 mg/m2 twice daily for 2 weeks followed by a 1 week rest period) in combination with docetaxel (75 mg/m2 as a 1 hour intravenous infusion every 3 weeks). 256 patients received docetaxel 100 mg/m2 alone.
TAXOTERE in combination with capecitabine resulted in statistically significant improvements in time to disease progression, overall survival and objective response rate compared to monotherapy with docetaxel as shown in the following table. Health related quality of life (HRQoL ) was assessed using the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaires (EORTC-QLQ), (C30 version 2, including Breast Cancer Module BR23). HRQoL was similar in the two treatment groups.
Breast cancer combination treatment efficacy results1
| Endpoint Parameter | Capecitabine/ docetaxel N=255 |
docetaxel N=256 |
Difference |
p-value |
|---|---|---|---|---|
| Time to Disease Progression | ||||
| median | 186 days | 128 days | HR2=0.643 | 0.0001 |
| [95% CI] | [165,198] | [105,136] | [0.563, 0.770] | |
| Survival | ||||
| median | 418 days | 338 days | HR2=0.753 | 0.0119 |
| [95% CI] | [374, 492] | [298, 362] | [0.603, 0.940] | |
| Response Rate | ||||
| 41.6 % | 29.7% | 11.9% | 0.0058 | |
| [95% CI] | [35.5, 47.9] | [24.2, 35.7] | [3.4, 20.0] |
1. All-randomised population, Investigator assessment
2. Hazard Ratio
TAXOTERE in combination with doxorubicin and cyclophosphamide: adjuvant therapy
Data from a multicentre open label randomized trial support the use of TAXOTERE for the adjuvant treatment of patients with node-positive breast cancer and KPS ≥ 80%, between 18 and 70 years of age. After stratification according to the number of positive lymph nodes (1-3, 4+), 1491 patients were randomized to receive either TAXOTERE 75 mg/m2 administered 1-hour after doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2 (TAC arm), or doxorubicin 50 mg/m2 followed by fluorouracil 500 mg/m2 and cyclophosphamide 500 mg/m2 (FAC arm). Both regimens were administered once every 3 weeks for 6 cycles. TAXOTERE was administered as a 1-hour infusion, all other drugs were given as IV bolus on day-1. G-CSF was administered in both arms as secondary prophylaxis to patients who experienced febrile neutropenia, prolonged neutropenia or neutropoenic infection. Patients in the Taxotere arm who continued to experience these reactions remained on G-CSF and had their dose reduced to 60 mg/m2. Patients on the TAC arm received antibiotic prophylaxis with ciprofloxacin 500 mg orally b.i.d. for 10 days starting on day 5 of each cycle, or equivalent. In both arms, after the last cycle of chemotherapy, patients with positive oestrogen and/or progesterone receptors received tamoxifen 20 mg daily for up to 5 years. Adjuvant radiation therapy was prescribed according to guidelines in place at participating institutions and was given to 69% of patients who received TAC and 72% of patients who received FAC.
An interim analysis was performed with a median follow up of 55 months. Significantly longer disease-free survival for the TAC arm compared to the FAC arm was demonstrated. In the TAC arm, 23% of subjects had experienced disease progression, compared to 30% in the FAC arm. TAC-treated patients had a 28% reduction in the risk of relapse compared to those treated with FAC (hazard ratio=0.72, 95% CI (0.59-0.88), p=0.001).
Overall survival was also significantly longer in the TAC arm with TAC-treated patients having a 30% reduction in the risk of death compared to FAC (hazard ratio= 0.70, 95% CI (0.53-0.91), p=0.008). In the TAC arm, 12% of patients had died compared to 17% on the FAC arm.
In the adjuvant breast cancer trial (TAX316), TAXOTERE in combination with doxorubicin and cyclophosphamide was administered to 744 patients of whom 48 (6%) were 65 years of age or greater. The number of elderly patients who received this regimen was not sufficient to determine whether there were differences in safety and efficacy between elderly and younger patients.
TAC-treated patient subsets according to prospectively defined major prognostic factors were analysed:
Analysis of TAC-treated patient subsets
| Disease Free Survival | Overall Survival | ||||||
|---|---|---|---|---|---|---|---|
| Patient subset | Number of patients | Hazard ratio* | 95% CI | P= | Hazard ratio* | 95% CI | P= |
| No of positive nodes | |||||||
| Overall | 745 | 0.72 | 0.59-0.88 | 0.001 | 0.70 | 0.53-0.91 | 0.008 |
| 1-3 | 467 | 0.61 | 0.46-0.82 | 0.0009 | 0.45 | 0.29-0.70 | 0.0002 |
| 4+ | 278 | 0.83 | 0.63-1.08 | 0.17 | 0.94 | 0.66-1.33 | 0.72 |
| Hormone Receptor status | |||||||
| Positive | 567 | 0.72 | 0.56-0.92 | 0.0076 | 0.69 | 0.48-1.00 | 0.0459 |
| Negative | 178 | 0.69 | 0.49-0.97 | 0.0296 | 0.66 | 0.44-0.98 | 0.0389 |
| Her-2 neu status | |||||||
| Positive | 155 | 0.60 | 0.41-0.88 | 0.0088 | 0.74 | 0.45-1.20 | 0.22 |
| Negative | 475 | 0.76 | 0.59-1.00 | 0.046 | 0.63 | 0.44-0.91 | 0.0135 |
*a hazard ratio of less than 1 indicates that TAC is associated with a longer disease-free survival and overall survival compared to FAC
The beneficial effect of TAC was seen in both hormone receptor positive and negative patients.
One phase II study was conducted in 20 previously untreated patients with locally advanced or metastatic non-small cell lung cancer. In this clinical trial, TAXOTERE was administered at a 75 mg/m2 dose given as a one-hour infusion every 3 weeks. The response rate was 10%.
Six phase II studies were conducted in patients with locally advanced or metastatic non-small cell lung cancer. A total of 160 patients had received no prior chemotherapy (previously untreated) and 88 patients had received prior platinum based chemotherapy (previously treated) which included 37 patients who had progressive disease with platinum therapy (platinum refractory). In these clinical trials, TAXOTERE was administered at a 100 mg/m2 dose given as a one-hour infusion every 3 weeks.
The 100 mg/m2 dose is associated with higher toxicity.
In the intent-to-treat analysis on previously untreated patients, the overall response rate (ORR) was 26.9% and in the previously treated population it was 17%. The survival time, for all previously untreated patients or previously treated patients, was 9 and 8 months respectively.
The safety and efficacy of TAXOTERE in patients with androgen independent (hormone refractory) metastatic prostate cancer were evaluated in a randomised multicentre Phase III trial. A total of 1006 patients with KPS ≥ 60 were randomised to the following treatment groups:
All 3 regimens were administered in combination with prednisone or prednisolone 5 mg twice daily, continuously.
Patients who received docetaxel every three weeks demonstrated significantly longer overall survival compared to those treated with mitozantrone (p=0.0094). The increase in survival seen in the docetaxel weekly arm was not statistically significant compared to the mitozantrone control arm. Efficacy endpoints for the TAXOTERE 3 weekly arm versus the control arm are summarised in the following table:
Efficacy of TAXOTERE in the Treatment of Patients with Androgen Independent (Hormone Refractory) Prostate Cancer (Intent-to-Treat Analysis)
| Endpoint | TAXOTERE Every 3 weeks |
Mitozantrone Every 3 weeks |
|---|---|---|
| Number of patients | 335 | 337 |
| Median survival (months) | 18.9 | 16.5 |
| 95% CI | (17.0-21.2) | (14.4-18.6) |
| Hazard ratio | 0.761 | - |
| 95% CI | (0.619-0.936) | - |
| p value+ * | 0.0094 | - |
| Number of patients | 291 | 300 |
| PSA** response rate (%) | 45.4 | 31.7 |
| 95% CI | (39.5-51.3) | (26.4-37.3) |
| p-value* | 0.0005 | - |
| Number of patients | 153 | 157 |
| Pain response rate (%) | 34.6 | 21.7 |
| 95% CI | (27.1-42.7) | (15.5-28.9) |
| p-value* | 0.0107 | - |
| Number of patients | 141 | 137 |
| Tumour response rate (%) | 12.1 | 6.6 |
| 95% CI | (7.2-18.6) | (3.0-12.1) |
| p-value* | 0.1112 | - |
+ stratified log rank test
* threshold for statistical significance=0.0175
**PSA : Prostate-Specific Antigen
TAXOTERE is indicated for the treatment of patients with locally advanced or metastatic breast cancer in whom previous chemotherapy has failed.
TAXOTERE in combination with capecitabine is indicated for the treatment of patients with locally advanced or metastatic breast cancer after failure of prior anthracycline containing chemotherapy.
TAXOTERE in combination with doxorubicin and cyclophosphamide is indicated for the adjuvant treatment of patients with node-positive breast cancer.
TAXOTERE is indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer, including those who have failed platinum-based chemotherapy.
TAXOTERE is indicated for the treatment of patients with androgen independent (hormone refractory) prostate cancer.
TAXOTERE is contraindicated in patients who have a history of severe hypersensitivity reactions to TAXOTERE or polysorbate 80.
TAXOTERE should not be used in patients with baseline neutrophil count of < 1.5 cells x 109/L.
TAXOTERE should not be used in patients with severe liver impairment.
TAXOTERE should not be used in pregnant or breast-feeding women.
Contraindications that apply for other drugs also apply when these drugs are combined with TAXOTERE.
The use of TAXOTERE should be confined to units specialised in the administration of cytotoxic chemotherapy and it should only be administered under the supervision of a qualified oncologist.
Patients should be pre-treated prior to each TAXOTERE administration. A premedication consisting of an oral corticosteroid such as dexamethasone 16 mg per day (e.g. 8 mg twice daily) for 3 days starting one day prior to TAXOTERE administration can reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reactions. (see Fluid retention and Hypersensitivity Reaction sections below, and refer to Dosage and Administration.)
For prostate cancer, the premedication is oral dexamethasone 8 mg, 12 hours, 3 hours and 1 hour before the docetaxel infusion.
Bone marrow suppression and other haematologic effects to TAXOTERE include neutropenia, the most frequent adverse reaction of TAXOTERE (see Adverse Effects, Clinical Studies).
Neutrophil nadirs occurred at a median of 7 days but this interval may be shorter in heavily pretreated patients. Frequent monitoring of complete blood counts should be conducted in all patients receiving docetaxel. Patients should be retreated with docetaxel only when neutrophils recover to a level ≥1.5 cells x 109/L.
TAXOTERE should not be administered to patients with baseline neutrophil counts of less than < 1.5 cells x 109/L. Frequent monitoring of complete blood counts should be conducted on all patients during treatment with docetaxel. Patients should not be retreated with TAXOTERE until neutrophils recover to a level ≥1.5 cells x 109/L. (see Dosage and Administration).
In the case of severe neutropenia (<0.5 cells x 109/L for seven days or more) during a course of TAXOTERE therapy, a reduction in dose for subsequent courses of therapy or the use of appropriate symptomatic measures are recommended. Prophylactic G-CSF may be used to mitigate the risk of haematological toxicities.
Patients who receive adjuvant therapy for breast cancer and who experience febrile neutropenia should receive G-CSF in all subsequent cycles. Patients who continue to experience this reaction should remain on G-CSF and have their TAXOTERE dose reduced (see also Dosage adjustments during treatment).
Patients should be observed closely for hypersensitivity reactions especially during the first and second infusions. Hypersensitivity reactions may occur within a few minutes, during or immediately following the cessation of the infusion of TAXOTERE, thus facilities for the treatment of hypotension and bronchospasm should be available. Frequently reported symptoms were flushing, rash with or without pruritus, chest tightness, back pain, dyspnoea and drug fever or chills. If hypersensitivity reactions occur, minor symptoms such as flushing or localised cutaneous reactions do not require interruption of therapy. However, severe reactions, such as severe hypotension, bronchospasm or generalised rash/erythema require immediate discontinuation of TAXOTERE and aggressive therapy. Severe symptoms are usually resolved after discontinuing the infusion and appropriate therapy. Patients who have developed severe hypersensitivity reactions should not be rechallenged with TAXOTERE.
Reversible cutaneous reactions were generally mild to moderate. Reactions were characterised by a rash including localised eruptions mainly on feet, hands (including severe hand and foot syndrome), but also arms, face or thorax, and frequently associated with pruritus. Eruptions generally occurred within one week after the docetaxel infusion. Less frequently, severe symptoms such as eruptions followed by desquamation which rarely led to interruption or discontinuation of docetaxel treatment where reported. Severe nail disorders were characterised by hypo- or hyperpigmentation, pain and onycholysis.
Very rare cases of cutaneous lupus erythematosus and bullous eruptions such as erythema multiform, Stevens-Johnson syndrome, toxic epidermal necrolysis, have been reported with docetaxel. In some cases multiple factors such as concomitant infections, concomitant medications and underlying disease may have contributed to the development of these effects.
Rare cases of ototoxicity, hearing disorders and/or hearing loss have been reported, including cases associated with other ototoxic drugs.
A premedication consisting of an oral corticosteroid such as dexamethasone 16 mg per day (e.g. 8 mg twice daily) for 3 days starting one day prior to docetaxel administration, unless contraindicated, can reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reactions (see Dosage and Administration).
Fluid retention is cumulative in incidence and severity, and is slowly reversible after docetaxel treatment is stopped. The median cumulative dose to onset for treatment with 75 mg/m2 is 524 mg/m2 and treatment at 100 mg/m2 is 509 mg/m2 (without premedication) and 797 mg/m2 (with premedication).
Patients with severe fluid retention such as pleural effusion, pericardial effusion and ascites should be monitored more closely.
Liver function tests (LFTs) should be measured at baseline and before each cycle.
In patients treated with docetaxel at 100 mg/m2 who have both elevations of serum transaminase values (ALT and /or AST) greater than 1.5 times the upper limit of normal (ULN) and increases in alkaline phosphatase greater than 2.5 times the ULN, there is a greater risk of developing severe adverse reactions such as toxic deaths including sepsis, gastrointestinal haemorrhage which can be fatal, febrile neutropenia, infections, thrombocytopenia, stomatitis and asthenia. The recommended dose of docetaxel in patients with elevated LFTs is 75 mg/m2 (see Dosage and Administration).
For those patients with increased serum bilirubin and/or values > 3.5 times the ULN for ALT and AST and 6 times the ULN for alkaline phosphatase, no dose-reduction can be recommended and docetaxel should not be used unless strictly indicated.
The development of severe neurosensory signs and/or symptoms have been observed in patients and requires a reduction of dose (see Dosage and Administration).
There have been no formal clinical studies to evaluate the drug interactions of docetaxel.
In vitro studies suggest that isoenzymes of the cytochrome P450 3A subfamily appear to be involved in the hepatic metabolism of docetaxel in humans. In vitro, the biotransformation of docetaxel was inhibited by cyclosporin, terfenadine, ketoconazole, erythromycin and troleandomycin and to a lesser extent by doxorubicin, vinorelbine, vinblastine and nifedipine, increased by dexamethasone, phenobarbitone and clofibrate and unaffected by cimetidine, ranitidine, omeprazole, diazepam, imipramine, paracetamol, caffeine, tolbutamide and quinidine. Strong P450 3A inhibitors may affect docetaxel metabolism in vivo, necessitating caution in co-administration regimens.
In vitro, plasma protein binding was more than 95%, with the important proteins being albumin, α1-acid glycoprotein and lipoproteins. The in vitro plasma protein binding of docetaxel was not affected by dexamethasone, erythromycin, salicylate, sulfamethoxazole, diphenhydramine, propranolol, propafenone, phenytoin and sodium valproate. The binding of digitoxin was not affected by docetaxel.
TAXOTERE was not mutagenic in bacterial or CHO/HPRT gene mutation assays, but was mutagenic in the in vitro chromosome aberration assay, in the in vivo micronucleus test in the mouse and modified the distribution of CHO-K1 cells in the cell cycle phases.
The carcinogenic potential of TAXOTERE has not been studied. However, based upon its pharmacodynamic mechanism of action, TAXOTERE may be a carcinogen.
Studies in mice have shown that IV doses of 144 mg/m2 or 30 mg/m2/day for 5 days are associated with testicular atrophy, mineralisation and degeneration of tubular germinal epithelium, Leydig cell hyperplasia, epididymal hypospermia, and follicular atresia in the ovaries. Studies in rats have shown that intravenous doses of 120 mg/m2 are associated with testicular atrophy, germ cell atrophy, Leydig cell hyperplasia and mineralisation. The rodent studies suggest that TAXOTERE may impair fertility. Studies in rats have also shown that IV doses of 0.9 mg/m2/day to both sexes are associated with reduced litter averages for corpora lutea, implantations and live foetuses, and increased litter averages for early and total resorptions. Larger doses to both sexes (males 1.8 mg/m2/day, females 1.35 mg/m2/day) are additionally associated with increased time to mating, increased number of dams with total resorption, and reduced male foetal body weight.
Docetaxel may cause foetal harm when administered to a pregnant woman.
Foetal radioactivity has been detected following intravenous (IV) administration of radiolabelled docetaxel to pregnant rats. Docetaxel has been shown to be embryo- and foetotoxic in rats and rabbits. At IV doses of 0.9 mg/m2, docetaxel caused fewer corpora lutea, fewer implantations, increased resorptions and embryofoetal deaths in rats. No evidence of teratogenic effects was found when docetaxel was administered IV at doses up to 1.8 mg/m2 or 1.2mg/m2 in rats or rabbits, respectively, but reduced foetal weight and delayed ossification were observed.
Offspring from rats receiving docetaxel 1.5 mg/m2/day IV from late gestation until weaning showed signs of delayed development. No studies have been performed in pregnant women.
If docetaxel is used during pregnancy, or if the patient becomes pregnant while receiving this drug, she should be appraised of the potential hazard. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with this drug. Contraceptive measures must be taken during and for at least three months after cessation of therapy with TAXOTERE.
Radioactivity has been detected in milk following intravenous administration of radiolabelled docetaxel to lactating rats. Offspring from rats receiving docetaxel 1.5 mg/m2/day IV during late gestation and lactation showed signs of delayed development. It is not known whether TAXOTERE is excreted in human milk. It is recommended to advise women not to breast-feed during treatment with TAXOTERE.
An analysis of safety data in patients equal to or greater than 60 years of age treated with TAXOTERE in combination with capecitabine showed an increase in the incidence of treatment-related grade 3 or 4 adverse reactions, treatment-related serious adverse reactions and early withdrawals from treatment due to adverse reactions compared to patients less than 60 years of age.
Of the 333 patients treated with TAXOTERE every three weeks in the prostate cancer study, 209 patients were 65 years of age or greater and 68 patients were older than 75 years. Differences in efficacy were not identified between elderly patients and younger patients. In patients treated with TAXOTERE every three weeks, the incidence of anaemia, infection, nail changes, anorexia, weight loss occurred at rates ≥ 10% higher in patients who were 65 years of age or greater compared to younger patients.
There are no data available in patients>70 years of age on TAXOTERE use in combination with doxorubicin and cyclophosphamide.
TAXOTERE is an antineoplastic agent and, as with other potentially toxic compounds, caution should be exercised when handling it and preparing TAXOTERE solutions. The use of gloves is recommended.
If TAXOTERE concentrate, premix solution or infusion solution comes into contact with the skin, wash immediately and thoroughly with soap and water. If TAXOTERE concentrate, premix solution or infusion solution comes into contact with mucous membranes, wash immediately and thoroughly with water.
The adverse reactions considered to be possibly or probably related to starting the administration of TAXOTERE have been obtained from 75 patients who received a dose of 75 mg/m2 without the recommended premedication, and from 2106 (2045 with normal* and 61 with elevated* LFTs at baseline) patients who received an initially planned dose of 100 mg/m2 over a one hour infusion every 3 weeks independently of the pre-medication. The patients were enrolled in 40 phase II and III studies conducted in Europe and North America (991 with breast carcinoma, 668 with non-small cell lung carcinoma and 447 with various tumour types).
The safety profile is generally similar between patients receiving TAXOTERE for the treatment of breast and non-small cell lung carcinoma.
The following table lists the adverse reaction data:
Summary of adverse events in patients receiving TAXOTERE at 75 and 100 mg/m2 as a single agent
| Normal LFTs* at Baseline | Elevated LFTs* at Baseline |
||
|---|---|---|---|
| TAXOTERE dosage | 75 mg/m2 | 100 mg/m2 | 100 mg/m2 |
| Number of patients | n=75 % |
n=2045 % |
n=61 % |
| Haematological Toxicity | |||
| Neutropenia | |||
| ANC~ <2.0 cells x 109/L | - | 95.5 | 96.4 |
| ANC~ <0.5 cells x 109/L | 73.0 | 75.4 | 87.5 |
| Febrile neutropenia | |||
| (fever/ANC~ <0.5 x 109/L): | |||
| by patient | - | 11.0 | 26.2 |
| by cycle | - | 2.6* | 8.7 |
| (fever/ANC~ <1 x 109/L): | |||
| by patient | 5.0 | - | - |
| by cycle | 1.5 | - | - |
| Thrombocytopenia | |||
| <100 cells x 109/L | 6.7 | 8.0 | 24.6 |
| Anaemia | |||
| <110 g/L | 86.7 | 90.4 | 91.8 |
| <80 g/L | 9.0 | 8.8 | 31.1 |
| Non-Haematological Toxicity | |||
| Body as a whole: | |||
| Fluid retention | |||
| Regardless of premedication: | |||
| All | 61.0 | 47.0 | 39.3 |
| Severe | 9.3 | 6.9 | 8.2 |
| 3 day premedication: | [n=92] | [n=3] | |
| All | 64.1 | 66.7 | |
| Severe | 6.5 | 33.3 | |
| Hypersensitivity reactions | |||
| Regardless of premedication: | |||
| All | 41.3 | 21.0 | 19.7 |
| Severe | 1.3 | 4.2 | 9.8 |
| 3 day premedication: | n=92 | n=3 | |
| All | 15.2 | 33.3 | |
| Severe | 2.2 | 0.0 | |
| Infections: | |||
| overall | 20.0 | 21.6* | 32.8 |
| Severe | 1.3 | 6.1 | 16.4 |
| Asthenia | |||
| All | 56.0 | 21.6* | 32.8 |
| Severe | 5.0 | 6.1 | 16.4 |
| Myalgia | 10.7 | 18.9 | 16.4 |
| Arthralgia | 0.0 | 9.2 | 6.6 |
| Neurological | |||
| Neurosensory: | |||
| All | 37.0 | 49.3 | 34.4 |
| Severe | 1.3 | 4.3 | 0.0 |
| Neuromotor | |||
| All | 4.0 | 13.8 | 6.6 |
| Severe | 0.0 | 3.6 | 1.6 |
| Cutaneous | |||
| Skin: | |||
| All | 45.3 | 47.6 | 54.1 |
| Severe | 1.3 | 4.8 | 9.8 |
| Nail disorders | 50.0 | 30.6 | 23.0 |
| Alopecia | 92.0 | 75.8 | 62.3 |
| Gastrointestinal | |||
| Nausea | 44.0 | 38.9 | 37.7 |
| Diarrhoea | 28.0 | 38.7 | 32.8 |
| Vomiting | 21.0 | 22.3 | 23.0 |
| Stomatitis | |||
| All | 10.7 | 41.7 | 49.2 |
| Severe | 2.6 | 5.5 | 13.0 |
| Mucositis | 40.0 | - | - |
| Infusion site reactions | 5.3 | 4.4 | 3.3 |
*Normal liver function tests (LFTs): transaminase ≤ 1.5 times upper limit of
normal or alkaline phosphatase
≤ 2.5 times upper limit of normal or isolated elevations of transaminase or
alkaline phosphatase up to 5 times upper limit of normal.
~ANC - Absolute neutrophil count
Thirty-five toxic deaths (1.7%) were reported in the 2045 patients with normal baseline liver function tests treated with TAXOTERE as monotherapy at the initially planned dose of 100mg/m2. Septic deaths (neutropenic infections, pneumonia or sepsis) accounted for 80% of the toxic deaths. The incidence of toxic deaths was higher (9.8%) in patients with elevated baseline LFTs.
Hypersensitivity reactions generally occur within a few minutes of the start of infusion and were generally mild to moderate. Frequently reported symptoms were flushing, rash with or without pruritus, chest tightness, back pain, dyspnoea and drug fever or chills (see Precautions).
Bone marrow suppression and other haematologic adverse reactions to docetaxel include:
Neutropoenia (in patients who did not receive G-CSF), the most frequent adverse reaction, was reversible and not cumulative. The median day to nadir was 7 days and the median duration of severe neutropoenia was 7 days.
Febrile neutropoenia and severe infections associated with neutrophil counts <0.5 x 109/L, infectious episodes (severe including sepsis pneumonia, fatal in 1.7%), occurred. Thrombocytopoenia, bleeding episodes (rarely associated with severe thrombocytopenia) and anaemia (severe) were also reported.
Mild to moderate neuro-sensory signs and/or symptoms occurred in 50% of the patients. Severe neurosensory symptoms (paresthenia, dysesthesia, pain including burning) were observed in 4.1% of metastatic breast cancer patients, and resulted in treatment discontinuation in 2%. Neuro-motor events (13.8% with 4% severe) mainly characterised by weakness. When these symptoms occur, dosage must be adjusted. If symptoms persist, treatment should be discontinued. Patients who experienced neurotoxicity in clinical trials and for whom follow-up information on the complete resolution of the event were available had spontaneous reversal of symptoms with a median of 81 days from onset (range 0 to 741 days).
Rare cases of convulsion or transient loss of consciousness have been observed with docetaxel administration. These reactions sometimes appear during the infusion of the drug.
In patients treated at 100mg/m2 as a single agent, increase in serum levels of AST, ALT, bilirubin and alkaline phosphatase greater than 2.5 the ULN were observed in less than 5% of patients. Very rare cases of hepatitis have been reported.
TAXOTERE in combination with capecitabine
The adverse reaction profile is consistent with the known toxicities of monotherapy treatments.
The most frequent treatment-related adverse reactions (≥ 5%) reported in the phase III clinical trial for TAXOTERE in combination with capecitabine in patients with locally advanced and/or metastatic breast cancer (n = 251) are shown in the table below.
The mean duration of treatment was 129 days in the combination arm and 98 days in the monotherapy arm. A total of 66 patients (26%) in the combination arm and 49 (20%) in the monotherapy arm discontinued from the trial because of adverse reactions. The percentages of patients requiring dose reductions due to adverse reactions were 65% in the combination arm and 36% in the monotherapy arm.
Treatment-related adverse reactions reported in ≥ 5% of patients treated with TAXOTERE in combination with capecitabine
| Capecitabine 1250 mg/m2 twice daily with TAXOTERE 75 mg/m2 /3 weeks (n=251) |
Docetaxel 100 mg/m2 /3 weeks (n=255) |
|||
|---|---|---|---|---|
| Body system Adverse reaction |
All Grades % |
Grade 3 / 4 % |
All Grades % |
Grade 3 / 4 % |
| Gastrointestinal | ||||
| Stomatitis | 67 | 18 | 42 | 5 |
| Diarrhoea | 64 | 14 | 45 | 5 |
| Nausea | 43 | 6 | 35 | 2 |
| Vomiting | 33 | 4 | 22 | 1 |
| Constipation | 14 | 1 | 12 | – |
| Abdominal pain | 14 | 2 | 9 | 1 |
| Dyspepsia | 12 | – | 5 | <1 |
| Abdominal pain upper | 9 | – | 6 | 1 |
| Dry mouth | 5 | – | 4 | – |
| Cutaneous | ||||
| Hand-foot syndrome | 63 | 24 | 7 | 1 |
| Alopecia | 41 | 6 | 42 | 7 |
| Nail disorder | 14 | 2 | 15 | – |
| Dermatitis | 8 | – | 9 | 1 |
| Rash erythematous | 8 | <1 | 4 | – |
| Nail discoloration | 6 | – | 4 | <1 |
| Onycholysis | 5 | 1 | 5 | 1 |
| General | ||||
| Asthenia | 23 | 3 | 22 | 5 |
| Pyrexia | 21 | 1 | 29 | <1 |
| Fatigue | 21 | 4 | 25 | 5 |
| Weakness | 13 | 1 | 9 | 2 |
| Pain in limb | 9 | <1 | 8 | <1 |
| Lethargy | 6 | – | 5 | 1 |
| Pain | 6 | – | 2 | – |
| Neurologic | ||||
| Taste disturbance | 15 | <1 | 14 | <1 |
| Paresthesia | 11 | <1 | 15 | 1 |
| Dizziness | 9 | – | 6 | <1 |
| Headache | 7 | <1 | 8 | 1 |
| Peripheral neuropathy | 5 | – | 10 | 1 |
| Cardiovascular | ||||
| Lower limb oedema | 14 | 1 | 12 | 1 |
| Sore throat | 11 | 2 | 7 | <1 |
| Dyspnoea | 7 | 1 | 9 | <1 |
| Cough | 6 | <1 | 9 | – |
| Epistaxis | 5 | <1 | 5 | – |
| Metabolism | ||||
| Anorexia | 12 | 1 | 10 | 1 |
| Decreased appetite | 10 | – | 4 | – |
| Dehydration | 8 | 2 | 5 | 1 |
| Decreased weight | 6 | – | 4 | – |
| Eye | ||||
| Increased lacrimation | 12 | – | 5 | – |
| Musculoskeletal | ||||
| Myalgia | 14 | 2 | 24 | 2 |
| Arthralgia | 11 | 1 | 18 | 2 |
| Back pain | 7 | 1 | 6 | 1 |
| Infection | ||||
| Oral candidiasis | 6 | <1 | 7 | <1 |
| Haematologic * | ||||
| Decreased haemoglobin | 13 | 4 | 11 | 4 |
| Neutropenia | 17 | 16 | 16 | 14 |
| Neutropenic fever | 21 | 16 | 21 | 21 |
| Leucopenia | 3 | 3 | 2 | 2 |
| Biochemical laboratory abnormalities * | ||||
| Increased alkaline phosphatase | 51 | 1 | 48 | 2 |
| Increased bilirubin | 23 | 9 | 6 | 3 |
| Increased AST | 42 | 3 | 37 | 4 |
| Increase ALT | 30 | 2 | 30 | 2 |
| Serum creatinine | 7 | <1 | 4 | - |
* Grades according to National Cancer Institute of Canada Toxicity Criteria, version 1, Dec 1994 were used.
Rare or uncommon adverse reactions, as described for capecitabine monotherapy, can be expected for combination therapy as well. Refer to capecitabine Product Information for adverse reactions which are at least remotely related to capecitabine occurring in < 5% of patients treated with capecitabine in combination with docetaxel.
The following table presents clinically important treatment-emergent adverse events (TEAEs) observed in 744 patients, who were treated with TAXOTERE 75 mg/m2 every 3 weeks in combination with doxorubicin and cyclophosphamide and 736 patients, treated with the comparator study drugs.
Clinically Important Treatment Emergent Adverse Events (TEAEs) considered related to study treatment in Patients Receiving TAXOTERE in Combination with Doxorubicin and Cyclophosphamide
| TAXOTERE 75 mg/m2 + Doxorubicin 50 mg/m2 +
Cyclophosphamide 500 g/m2 n=744 |
Fluorouracil 500 mg/m2 + Doxorubicin 50 mg/m2 +
Cyclophosphamide 500 g/m2 n=736 |
|||
|---|---|---|---|---|
| Body system Adverse reaction |
All Grades (%) |
Grade 3 / 4 (%) |
Any (%) |
Grade 3 / 4 (% |
| Cutaneous | ||||
| Alopecia | 97.7 | N/A | 97.1 | N/A |
| Skin toxicity | 18.4 | 0.7 | 10.9 | 0.3 |
| Nail disorder | 18.4 | 0.4 | 13.9 | 0.1 |
| Haematologic | ||||
| Anaemia | 91.5 | 4.3 | 71.7 | 1.6 |
| Neutropenia | 71.4 | 65.5 | 82.0 | 49.3 |
| Thrombocytopenia | 39.4 | 2.0 | 27.7 | 1.2 |
| Febrile neutropenia | 24.7 | N/A | 2.5 | N/A |
| Neutropoenic infection | 12.1 | N/A | 6.3 | N/A |
| Gastrointestinal | ||||
| Nausea | 80.4 | 5.1 | 87.4 | 9.5 |
| Stomatitis | 69.1 | 7.1 | 52.6 | 2.0 |
| Vomiting | 42.6 | 4.3 | 58.2 | 7.3 |
| Diarrhoea | 30.9 | 3.2 | 23.5 | 1.0 |
| Constipation | 22.6 | 0.4 | 21.5 | 1.2 |
| Abdominal pain | 7.3 | 0.5 | 3.3 | 0.0 |
| General | ||||
| Asthenia | 79.2 | 11.0 | 69.4 | 5.2 |
| Fever in absence of infection | 43.1 | 1.2 | 13.2 | 0.0 |
| Infection* | 27.2 | 3.2 | 17.4 | 1.4 |
| Peripheral oedema | 26.7 | 0.4 | 7.2 | 0.0 |
| Hypersensitivity reactions | 10.5 | 1.1 | 2.2 | 0.0 |
| Lymphoedema | 0.3 | 0.0 | 0.0 | 0.0 |
| Gynaceologic | ||||
| Amenorrhoea | 57.6 | N/A | 48.1 | N/A |
| Neurologic | ||||
| Taste Perversion | 27.4 | 0.7 | 15.1 | 0.0 |
| Neuorpathy sensory | 23.8 | 0.0 | 7.9 | 0.0 |
| Neuro-cortical | 2.8 | 0.3 | 3.9 | 0.3 |
| Neuropathy motor | 2.8 | 0.0 | 1.5 | 0.0 |
| Neuro-cerebellar | 51.1 | –0.1 | 0.8 | 0.0 |
| Syncope | 0.5 | 0.0 | 0.4 | 0.0 |
| Musculoskeletal | ||||
| Myalgia | 22.8 | 0.8 | 8.0 | 0.0 |
| Arthralgia | 15.1 | 0.4 | 5.7 | 0.3 |
| Cardiovascular | ||||
| CHF | 0.0 | 1.6 | 0.0 | 0.5 |
| Vasodilatation | 20.3 | 0.9 | 15.9 | 0.4 |
| Cardiac dysrhythmias** | 3.9 | 0.1 | 2.9 | 0.3 |
| Hypotension | 1.5 | 0.0 | 0.5 | 0.0 |
| Phlebitis | 0.7 | 0.0 | 0.4 | 0.0 |
| Metabolic | ||||
| Anorexia | 19.9 | 2.2 | 16.4 | 1.2 |
| Weight gain or loss | 15.2 | 0.3 | 9.2 | 0.0 |
| Eye | ||||
| Lacrimation disorder | 9.8 | 0.1 | 6.4 | 0.0 |
| Conjunctivitis | 4.6 | 0.3 | 6.0 | 0.1 |
| Respiratory | ||||
| Cough | 3.1 | 0.0 | 2.2 | 0.1 |
N/A: not applicable.
* there was no septic death in either treatment arms.
** one patient died due to heart failure in TAC arm
Of the 744 patients treated with TAC, 33.1% experienced severe TEAEs. Dose reductions due to haematologic toxicity occurred in 1% of cycles in TAC arm. Six percent of patients treated with TAC discontinued treatment due to adverse events; fever in the absence of infection and allergy being the most common reasons for withdrawal.
In addition to gastrointestinal events reflected in the above table, 4 patients were reported to have colitis/ enteritis/ large intestine perforation in the TAC arm. Two of these patients required treatment discontinuation; no deaths due to these events occurred.
Acute Myeloid Leukaemia/ Myelodysplastic Syndrome
At a median follow-up time of 55 months, 3 patients were diagnosed with leukaemia (2 in TAC versus 1 in FAC arm). An additional case of leukaemia was reported in the TAC arm after the follow-up period. No cases of myelodysplastic syndrome occurred.
The adverse reaction profile is consistent with the known safety profile of TAXOTERE.
The table below provides the percentage of subjects with clinically important treatment-emergent adverse events (TEAEs) and haematological toxicities related to study treatment, reported in the phase III clinical trial for TAXOTERE 75 mg/m2 q3w and mitozantrone q3w in combination with prednisone (or prednisolone).
Clinically Important Treatment-Emergent Adverse Events Related to Study Medication.
| TAXOTERE 75 mg/m2 every 3 weeks (n=322) | Mitozantrone 12 mg/m2 every 3 weeks (n=335) | |||
|---|---|---|---|---|
| Grade 3/4 | Any | Grade 3/4 | Any | |
| Cutaneous | ||||
| Alopecia | N/A* | 65.1 | N/A* | 12.5 |
| Nail changes | 0.0 | 28.3 | 0.0 | 6.6 |
| Rash/desquamation | 0.3 | 3.3 | 0.0 | 0.9 |
| Haematologic | ||||
| Neutropenia | 32.0 | 40.9 | 21.7 | 48.2 |
| Anaemia | 1.9 | 66.5 | 1.8 | 57.8 |
| Thrombocytopenia | 0.6 | 3.4 | 1.2 | 7.8 |
| Epistaxis | 0.0 | 3.0 | 0.0 | 0.6 |
| Febrile neutropenia | N/A | 2.7 | N/A* | 1.8 |
| General | ||||
| Fatigue | 3.9 | 42.8 | 2.7 | 26.6 |
| Infection | 3.3 | 12.0 | 2.1 | 4.8 |
| Stomatitis/pharyngitis | 0.9 | 17.8 | 0.0 | 7.8 |
| Fluid retention | 0.6 | 24.4 | 0.3 | 4.5 |
| Allergic reaction | 0.6 | 6.9 | 0.0 | 0.3 |
| Anorexia | 0.6 | 12.7 | 0.0 | 11.6 |
| Gastrointestinal | ||||
| Nausea | 2.4 | 35.5 | 0.9 | 28.7 |
| Diarrhoea | 1.2 | 24.1 | 0.9 | 4.2 |
| Vomiting | 1.2 | 13.3 | 0.6 | 7.2 |
| Neurologic | ||||
| Neuorpathy sensory | 1.2 | 27.4 | 0.0 | 2.1 |
| Taste disturbance | 0.0 | 17.5 | 0.0 | 6.3 |
| Neuropathy motor | 0.0 | 3.9 | 0.0 | 0.9 |
| Respiratory | ||||
| Dyspnea | 0.6 | 4.5 | 0.3 | 3.3 |
| Cough | 0.0 | 1.2 | 0.0 | 0.9 |
| Eye | ||||
| Tearing | 0.6 | 9.3 | 0.0 | 1.5 |
| Musculoskeletal | ||||
| Myalgia | 0.3 | 6.9 | 0.0 | 3.3 |
| Arthralgia | 0.3 | 3.0 | 0.0 | 0.6 |
| Cardiovascular | ||||
| Abnormal cardiac left ventricular function | 0.3 | 3.9 | 0.9 | 19.1 |
*NA: not applicable.
The following information relates to serious events observed following the marketing of TAXOTERE. Voluntary reports of serious adverse events that have been received since market introduction (without causal relationship) that are not listed previously are cited below. Frequency estimates are as follows: common ? 1- 10%, uncommon 0.1-1%; rare 0.01-0.1%; very rare <0.01%.
Body as a whole
Uncommon: chest pain, diffuse pain.
Rare: abdominal pain.
Very rare: radiation recall phenomenon.
Cutaneous
Very rare: bullous eruption such as erythema multiforme or Stevens-Johnson syndrome. Multiple factors such as concomitant infections, concomitant medications and underlying disease may have contributed to the development of these effects.
Severe nail disorders characterised by hypo- or hyperpigmentation, and infrequently onycholysis and pain.
Gastrointestinal
Rare: constipation, oesophagitis and taste perversion, gastrointestinal haemorrhage, dehydration as a consequence of gastrointestinal events, ileus and intestinal obstruction.
Very rare: duodenal ulcer, gastrointestinal perforation, neutropenic enterocolitis, colitis including ischemic colitis.
Neurologic
Rare: confusion, seizures, transient loss of consciousness. These reactions sometimes occur during infusion of the drug.
Cardiovascular
Common: hypertension, hypotension.
Uncommon: cardiac arrhythmia, congestive heart failure.
Rare: atrial fibrillation, syncope, tachycardia.
Very rare: deep vein thrombosis, myocardial infarction, ECG abnormalities, thrombophlebitis, pulmonary embolism.
Hepatic
Very rare: hepatitis, sometimes fatal, primarily in patients with pre-existing liver disorders, have been reported.
Respiratory
Uncommon: dyspnoea.
Rare: Acute respiratory distress syndrome, interstitial pneumonia, acute oedema,.pulmonary fibrosis and radiation recall pneumonia. Rare cases of radiation pneumonitis have been reported in patients receiving concomitant radiotherapy.
Very rare: pulmonary fibrosis.
Urogenital
Rare: renal insufficiency.
Common: generalised or localised pain including chest pain without cardiac or respiratory involvement.
Rare cases of lacrimation with or without conjunctivitis have been reported and very rare cases of lacrimal duct obstruction resulting in excessive tearing have been reported primarily in patients receiving other anti-tumour agents concomitantly.
Rare cases of transient visual disturbances (flashes, flashing lights, scotomata) typically occurring during drug infusion and in association with hypersensitivity have been reported. These were reversible upon discontinuation of the infusion.
The recommended dosage of TAXOTERE is 75 to 100 mg/m2 administered as a one-hour infusion every three weeks (see Preparation for the Intravenous Administration). A dose of 100 mg/m2 has been shown to result in a moderate increase in response rates compared with 75 mg/m2 but is associated with greater toxicity.
The recommended dose of TAXOTERE in the adjuvant treatment of breast cancer is 75 mg/m2 administered 1 hour after doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2 every 3 weeks for a total of six cycles (see also Dosage adjustments during treatment and PRECAUTIONS, Haematology).
The recommended dosage of TAXOTERE is 75 mg/m2 administered as a one-hour infusion every three weeks when combined with capecitabine administered orally at 1,250 mg/m2 twice daily (within 30 minutes after the end of a meal) for two weeks followed by a 1 week rest period, given as 3 week cycles. Refer to capecitabine Product Information for capecitabine dose calculation according to body surface area.
The recommended dosage of TAXOTERE for prostate cancer is 75 mg/m2 administered as a one-hour infusion every three weeks. Prednisone or prednisolone 5 mg orally twice daily is administered continuously, commencing day 1 and continuing through each cycle.
A premedication consisting of an oral corticosteroid, such as dexamethasone 16 mg per day (e.g. 8 mg twice daily) for 3 days starting one day prior to docetaxel administration, unless contraindicated, can reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reactions.
For prostate cancer, given the concurrent use of prednisone or prednisolone, the recommended premedication regimen is oral dexamethasone 8 mg, 12 hours, 3 hours and 1 hour before the docetaxel infusion.
TAXOTERE should be administered when the neutrophil count is ≥ 1.5 cells x 109/L.
In patients treated at 75 mg/m2
Patients who experienced either febrile neutropenia, neutrophil < 0.5 cells x 109/L for more than one week, severe or cumulative cutaneous reactions or severe neurosensory signs and/or symptoms during TAXOTERE therapy should have the dosage of TAXOTERE reduced from 75 mg/m2 to 55 mg/m2(or to 60 mg/m2 for adjuvant therapy for breast cancer). If the patient continues to experience these reactions at 55 mg/m2 (or at 60 mg/m2), the treatment should be discontinued.
In patients treated at 100 mg/m2
Patients who experienced either febrile neutropenia, neutrophil < 0.5 cells x 109/L for more than one week, severe or cumulative cutaneous reactions or severe neurosensory signs and/or symptoms during TAXOTERE therapy should have the dosage of TAXOTERE reduced from 100 mg/m2 to 75 mg/m2. If the patient continues to experience these reactions at 75 mg/m2, the dosage should either be decreased from 75 mg/m2 to 55 mg/m2, or the treatment should be discontinued.
For capecitabine dose modifications when combined with TAXOTERE, see capecitabine Product Information.
For patients developing the first appearance of a Grade 2 toxicity which persists at the time of the next TAXOTERE/ capecitabine treatment, delay treatment until resolved to Grade 0-1, and resume at 100% of the original dose.
For patients developing the second appearance of Grade 2 toxicity, or the first appearance of a Grade 3 toxicity, at any time during the treatment cycle, delay treatment until resolved to Grade 0-1, then resume treatment with TAXOTERE 55 mg/m2.
For any subsequent appearances of toxicities, or any Grade 4 toxicities, discontinue the TAXOTERE dose.
In women receiving TAXOTERE, doxorubicin and cyclophosphamide (TAC), the risk of acute myeloid leukaemia is comparable to the risk observed for other anthracycline/cyclophosphamide containing regimens.
Patients who receive adjuvant therapy for breast cancer and who experience febrile neutropenia should receive G-SF in all subsequent cycles. Patients who continue to experience this reaction should remain on G-CSF and have their TAXOTERE dose reduced to 60mg/m2. If G-CSF is not used, the TAXOTERE dose should be reduced from 75 to 60mg/m2. Patients who experience Grade 3 or 4 stomatitis should have their dose decreased to 60mg/m2.
Patients with hepatic impairment
In patients treated at 75 mg/m2
For those patients with increased serum bilirubin and/or values > 3.5 times the ULN for ALT and AST and > 6 times the ULN for alkaline phosphatase, no dose-reduction can be recommended and docetaxel should not be used unless strictly indicated.
In patients treated at 100 mg/m2
Based on the pharmacokinetic data, in patients who have both elevations of transaminase values [ALT and/or AST greater than 1.5 times the upper limit of normal range (ULN)] and increases in alkaline phosphatase greater than 2.5 times the ULN, the recommended dose of docetaxel is 75 mg/m2 (see Pharmacokinetic Properties). For those patients with increased serum bilirubin and/or values > 3.5 times the ULN for ALT and AST and > 6 times the ULN for alkaline phosphatase, no dose-reduction can be recommended and docetaxel should not be used unless strictly indicated.
Children
The safety and effectiveness of TAXOTERE in children have not been established.
Elderly
Based on the population pharmacokinetics, there are no special instructions for the use in elderly.
For capecitabine dosage reduction when combined with TAXOTERE, see capecitabine Product Information.
TAXOTERE 20 mg concentrate and solvent vials
TAXOTERE 20 mg concentrate vial contains 0.5 ml of a 40 mg/ml solution of docetaxel in polysorbate 80 (fill volume: 24.4mg/ 0.61 ml). This fill volume has been established during the development of TAXOTERE to compensate for liquid loss during preparation of the premix solution due to foaming, adhesion to the walls of the vial, and the "dead volume". This overfill ensures that after dilution with the entire contents of the accompanying TAXOTERE Solvent vial, there is an initial diluted solution of 2 mL containing 10 mg/mL docetaxel which correspond to the labelled amount of 20 mg per vial.
Solvent vial contains 1.5 ml of a 13% w/w solution of ethanol in water for injections (fill volume: 1.98 ml). The addition of the entire contents of the solvent vial to the content of the TAXOTERE 20 mg concentrate vial ensures a premix concentration of 10 mg/ml docetaxel.
TAXOTERE 80 mg concentrate and solvent vials
TAXOTERE 80 mg concentrate vial contains 2 ml of a 40 mg/ml solution of docetaxel in polysorbate 80 (fill volume: 94.4mg/ 2.36 ml). This fill volume has been established during the development of TAXOTERE to compensate for liquid loss during preparation of the premix solution due to foaming, adhesion to the walls of the vial, and the "dead volume". This overfill ensures that after dilution with the entire contents of the accompanying TAXOTERE Solvent vial, there is an initial diluted solution of 8 mL containing 10 mg/mL docetaxel which correspond to the labelled amount of 80 mg per vial.
Solvent vial contains 6 ml of a 13% w/w solution of ethanol in water for injections (fill volume: 7.33 ml). The addition of the entire contents of the solvent vial to the content of the TAXOTERE 80 mg concentrate vial ensures a premix concentration of 10 mg/ml docetaxel.
a) Preparation and storage of the TAXOTERE premix solution (10 mg docetaxel/mL)
Using a syringe fitted with a needle, aseptically withdraw the ENTIRE contents of the TAXOTERE Solvent vial by partially inverting the vial. Inject the entire contents of the syringe into the corresponding TAXOTERE vial. Remove the syringe and needle and mix the vial by repeated inversions for at least 45 seconds. Do not shake. Allow the TAXOTERE premix solution to stand for 5 minutes at room temperature and then check that the solution is homogenous and clear. Foaming is normal even after 5 minutes due to the presence of polysorbate 80 in the formulation.
The premix solution contains 10 mg/ml docetaxel. As with all parenteral products, TAXOTERE premix solution should be visually inspected prior to use. Solutions containing a precipitate should be discarded. The premix solution should be used immediately after preparation. The chemical and physical stability of the premix solution has been demonstrated for 8 hours when stored either in the refrigerator or at room temperature. However, if storage is necessary, store the premix in the refrigerator to reduce microbiological hazards.
b) Preparation and storage of the infusion solution
More than one premix vial may be necessary to obtain the required dose for the patients. Based on the required dose for the patient expressed in mg, aseptically withdraw the corresponding premix volume containing 10 mg/mL docetaxel from the appropriate number of premix vials using a graduated syringe fitted with a needle. For example, a dose of 140 mg docetaxel would require 14mL docetaxel premix solution.
Inject the required premix volume into a 250 mL infusion bag or glass bottle containing either 0.9% sodium chloride solution or 5% glucose solution. If a dose greater than 200 mg of docetaxel is required, use a larger volume of the infusion vehicle so that a concentration of 0.74 mg/mL docetaxel is not exceeded. Mix the infusion bag or glass bottle manually using a rocking motion.
As with all parenteral products, TAXOTERE solution for infusion should be visually inspected prior to use, and solutions containing a precipitate should be discarded.
TAXOTERE solution for infusion should be aseptically administered intravenously as a one-hour infusion under room temperature and normal lighting conditions. The solution for infusion is stable at room temperature (15-25°C) for up to 8 hours (up to 24 hours in glass bottles). However to reduce microbiological hazards and the risk of crystallisation of docetaxel from diluted solutions, it is recommended that reconstitution and further dilution should be effected immediately prior to use and infusion commenced as soon as practicable after preparation of the mixture. Any residue after infusion should be discarded. Any solutions which are discoloured, hazy or contain visible particulate matter should not be used.
c) Disposal
All materials that have been utilised for dilution and administration should be disposed of according to standard procedures.
In case of overdosage, the patient should be kept in a specialised unit and vital functions closely monitored. There is no known antidote for TAXOTERE overdosage. The primary anticipated complications of overdosage would consist of bone marrow suppression, peripheral neurotoxicity and mucositis.
There were two reports of overdose. One patient received 150 mg/m2 and the other received 200 mg/m2 of TAXOTERE as a one hour infusion. They both recovered after experiencing severe neutropenia, mild asthenia, cutaneous reactions and mild paraesthesia.
TAXOTERE concentrate for infusion to be diluted is supplied in single-dose vials as a sterile pyrogen-free non-aqueous clear yellow to brown yellow viscous solution with the accompanying sterile solvent (13% ethanol in water for injections). The following strengths are available:
TAXOTERE® 20 mg
Each blister carton contains one single-dose TAXOTERE (docetaxel) parenteral preparation to be diluted equivalent to 20 mg docetaxel (anhydrous) in 0.5 mL polysorbate 80 (Fill: 24.4 mg / 0.61 mL) and one single-dose TAXOTERE® Solvent vial containing 1.98 mL 13% ethanol in water for injections.
http://www.medsafe.govt.nz/profs/datasheet/t/Taxotereinf.htm